Review Article
Management of Hematologic Adverse Events Associated With Immune Checkpoint Inhibitors
Barbara Barnes Rogers, CRNP, MN, AOCN®, ANP-BC, Carolyn Zawislak, MPAS, PA-C, and Victoria Wong, PA-C
From Fox Chase Cancer Center, Philadelphia, Pennsylvania
Authors’ disclosures of conflicts of interest are found at the end of this article.
Correspondence to: Barbara Barnes Rogers, CRNP, MN, AOCN®, ANP-BC, Fox Chase Cancer Center, 333 Cottman Avenue, Philadelphia PA 19111. E-mail: barbara.rogers@tuhs.temple.edu
J Adv Pract Oncol 2021;12(4):392–404 |
https://doi.org/10.6004/jadpro.2021.12.4.4 |
© 2021 Harborside™

 ABSTRACT
ABSTRACT
Immune checkpoint inhibitors target suppressor receptors, including cytotoxic T-lymphocyte–associated antigen 4 (CTLA-4), programmed cell death protein 1 (PD-1), and programmed cell death ligand 1 (PD-L1). The activated T cells are not antigen specific; therefore, the blockade of the immune checkpoint may result in the development of autoimmune adverse events. The most common immune-related adverse events (irAEs) are rash, colitis, and endocrinopathies. However, irAEs that affect the hematologic system are rare and can affect red blood cells (e.g., autoimmune hemolytic anemia), white blood cells, and platelets (e.g., immune thrombocytopenia). Usually one cell line is affected; however, in some cases, multiple cell lines can be affected. Other changes in the hematologic system can also be affected (e.g., cryoglobulinemia, cytokine release syndrome). Due to the rarity and lack of recognition of these AEs, the timing, spectrum of events, and clinical presentation are poorly understood. Management of hematologic irAEs usually involves the use of steroids; however, other agents (e.g., IVIG, cyclosporine, rituximab) or procedures (e.g., plasma exchange, transfusions) can also be used.
ARTICLE
Immune checkpoint inhibitors (ICIs) target suppressor receptors, including cytotoxic T-lymphocyte–associated antigen 4 (CTLA-4) and programmed cell death protein 1 (PD-1), which are located on the surface of immune cells. Since the activated T cells are not antigen specific, blockade of the immune checkpoint may result in the development of autoimmune adverse events (Leaf et al., 2019). Common immune-related adverse events (irAEs) include rash, colitis, and endocrinopathies.
In a recent review of hematologic irAEs secondary to PD-L1 inhibitors, the reported incidences of cytopenias (of all grades) with single-agent PD-L1 inhibition were as follows: anemia (5%), thrombocytopenia (2%), leukopenia (2%), and neutropenia (1%). The incidence of each cytopenia increased with combination therapy (Sui et al., 2018). Hematologic irAEs primarily impact blood counts within one cell line (i.e., neutropenia, thrombocytopenia) or more than one cell line (i.e., bicytopenia, pancytopenia) but may have other changes in the hematologic system (e.g., cryoglobulinemia, cytokine release syndrome). Specific hematologic irAEs that have been reported include autoimmune hemolytic anemia (AIHA), aplastic anemia (AA), pure red cell aplasia (PRCA), cold agglutinin syndrome (CAS), neutropenia, immune thrombocytopenia (ITP), acquired thrombotic thrombocytopenia purpura (TTP), bicytopenia or pancytopenia, hemophagocytic lymphohistiocytosis, myelodysplastic syndrome (MDS), post-transfusion purpura (PTP), and agranulocytosis (Davis et al., 2019; Michot et al., 2019; Noseda et al., 2019).
In a study by Davis and colleagues (2019), the hematologic irAEs were most commonly noted in individuals with melanoma and lung cancer. Davis and colleagues (2019) also noted that the median age of onset differed by specific hematologic toxicity but ranged from 59 to 66 years. Some patients who develop hematologic irAEs may develop a second hematologic irAE, most commonly AIHA and ITP (Davis et al., 2019). Hematologic irAEs can be fatal (Davis et al., 2019). The median time to onset of hematologic irAEs associated with CTLA-4 inhibitors was 40 days (either alone or in combination with PD-1 inhibitors) and occurred earlier than those associated with PD-1 or PD-L1 inhibitors. Due to the rarity of these toxicities and lack of recognition of these AEs, the timing, spectrum of events, and clinical presentation of hematologic events are poorly understood (Davis et al., 2019).
Anemias
Autoimmune Hemolytic Anemia (AIHA)
Autoimmune hemolytic anemia is a rare event associated with ICIs and involves the formation of autoantibodies against red blood cells (IgG, IgM, or both), leading to a marked decrease in the lifespan of red blood cells (Roumier et al., 2014). While rare, it is considered to be the most common hematologic irAE (Davis et al., 2019). Autoimmune hemolytic anemia is noted to occur more frequently with PD-1 or PD-L1 inhibitors (0.15%–0.25%) as compared with CTLA-4 inhibitors (0.6%; Tanios et al., 2018). Due to the increasing use of immunotherapy, the number of reports of ICI-associated AIHA has been increasing (Khan et al., 2017). Cases of AIHA from ICIs can be serious, with 15% of cases being fatal (Davis et al., 2019). Reported episodes of AIHA are most common in patients with malignant melanoma, then lung cancer, Hodgkin lymphoma, and renal cell cancer. Approximately 17.6% of cases were noted in patients with melanoma who were receiving combination therapy with a PD-1 inhibitor along with ipilimumab (Yervoy). Approximately 26.5% of cases also experienced other irAEs (Tanios et al., 2018). Tanios and colleagues (2018) reviewed the public U.S. Food & Drug Administration (FDA) databases and reported on the 68 cases of AIHA that were within the database. They indicated that AIHA can occur either early or later after the administration of an ICI, with median time to occurrence of 10 weeks (range: 2–78 weeks). The dose of the ICI does not seem to be related to the development of AIHA (Palla et al., 2016).
Two forms of drug-induced AIHA exist: warm AIHA and cold AIHA. These are dependent on the temperature of the autoantibodies that become active (Palla et al., 2016). Warm AIHA is usually mediated by IgG, and the autoantibodies are active at temperatures greater than 37°C. Cold AIHA results from IgM, and the autoantibodies are active at temperatures of 0 to 4°C. Autoimmune hemolytic anemia associated with ICIs is usually the warm type (Tanios et al., 2018). Patients with active AIHA are not considered candidates for concurrent ICI therapy; however, this issue has not been investigated (Tanios et al., 2018). It has been noted in two series that approximately 25% of patients who developed ICI-associated AIHA had an underlying lymphoproliferative disorder (e.g., chronic lymphocytic leukemia), as well as other conditions, including viral infections, babesiosis, and a prior allogeneic stem cell transplantation (Tanios et al., 2018). This phenomenon prompts the question of whether patients who have baseline immune dysfunction may be predisposed to ICI-associated AIHA (Leaf et al., 2019).
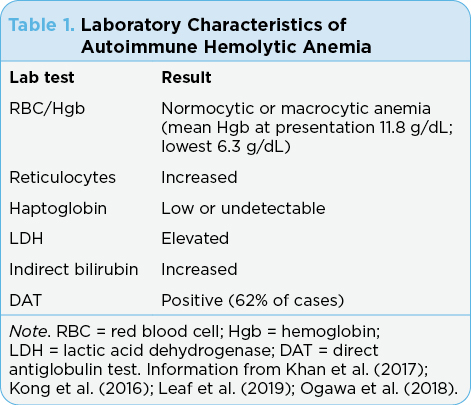
The diagnosis of AIHA is based on laboratory findings, including red blood cell/hemoglobin, reticulocyte count, haptoglobin level, lactate dehydrogenase (LDH), indirect bilirubin level, and direct antiglobulin test (DAT; see Table 1). There is a question if DAT negative and DAT positive are two distinct conditions (Leaf et al., 2019). Warm AIHA is usually associated with irAEs since most are DAT positive (Kong et al., 2016). The severity of hemolysis did not appear to differ based on the DAT status (Leaf et al., 2019).
Treatment guidelines for AIHA have not yet been fully developed but usually include immunosuppression resulting in the reduction of the production of autoantibodies (Table 2). Glucocorticoids are considered first line and most patients respond to steroids. However, at times, alternate therapies are needed such as intravenous immunoglobulin (IVIG) blood transfusions, hematopoietic growth factors, and other supportive treatments (Gnanapandithan et al., 2019). The treatment that is best for second-line therapy is currently unknown. While the presence of significant anemia (i.e., Hgb < 8.0 g/dL) might indicate the need to permanently discontinue ICIs, it has been noted that some patients can be retreated with the ICI and not develop recurrence of AIHA (Leaf et al., 2019). However, because hematologic AEs associated with ICIs are uncommon, the number of cases that have been successfully rechallenged is small. Therefore, there are no guidelines as to how to decide if patients are likely to be rechallenged successfully.


Cold Agglutinin Syndrome (CAS)
Cold antibodies account for approximately 25% of autoimmune hemolytic anemias (Berentsen et al., 2015). Cold agglutinins are antibodies that bind to erythrocyte surface antigens at low temperatures, causing agglutination and complement-mediated hemolysis (Berentsen et al., 2015). Cold agglutinin syndrome is different from cold agglutinin disease. Cold agglutinin disease is a clonal B-cell lymphoproliferative disorder, while cold agglutinin syndrome is similar but is a secondary cold hemolytic syndrome that arises from infections (i.e., Mycoplasma pneumoniae pneumonia or Epstein-Barr virus infection) or malignancies, and has been reported to be associated with ICIs. The development of CAS associated with ICIs is not related to duration or the dose of the ICI, nor is it related to the disease being treated by the ICI (Hasanov et al., 2018).
Direct antiglobulin test, peripheral blood smear features, bone marrow biopsy, and flow cytometry results assist in making the diagnosis of CAS. Bone marrow biopsy is expected to have increased cellularity and increased elements of all three lineages, but with no evidence of myelodysplastic syndrome, lymphoma, or myelodysplastic process.
Spontaneous remission occurs in nearly all cases of CAS, but there are no evidence-based guidelines in the management of CAS associated with ICI (Table 2). Interventions include packed red blood cell transfusions. Plasmapheresis can be considered in extreme cases (Berentsen et al., 2015). While steroids are the backbone of guidelines in the management of ICI-associated AEs, steroids have been found to occasionally be effective in managing primary CAS. Rituximab (Rituxan) has been noted to be highly effective in this setting and is expected to be helpful in managing ICI-associated CAS (Hasanov et al., 2018).
Pure Red Cell Aplasia (PRCA)
Pure red cell aplasia is a rare blood disease that is characterized by normocytic, normochromic anemia, reticulocytopenia, and the absence of hemoglobin-containing cells in an otherwise normal bone marrow aspirate. There are two types of PRCA: congenital (Diamond-Blackfan anemia) and acquired (Yuki et al., 2017). Acquired PRCA is associated with drugs including ICIs, infections, autoimmune diseases, and neoplasms. Pure red cell aplasia pathogenesis is not clearly understood but most likely has an autoimmune etiology; however, the production of antibodies against erythroid progenitors or erythropoietin has been noted.
Management of PRCA includes the use of immunosuppressive therapies such as corticosteroids, T-cell–directed agents (e.g., cyclosporine), and IVIG (Gordon et al., 2009; Table 2). The 10-year survival of patients with PRCA is 80% or more; however, it can be life threatening. Pure red cell aplasia often recurs as the dose of the steroid is tapered or discontinued. Therefore, long-term therapy is indicated (Yuki et al., 2017).
Thrombocytopenia
Immune Thrombocytopenia (ITP)
Immune thrombocytopenia is caused by the development of autoantibodies against platelets, usually after an inciting event of the immune system (Shiuan et al., 2017). It is a diagnosis of exclusion and is defined as isolated thrombocytopenia without associated anemia or leukopenia and without any other obvious cause for thrombocytopenia. Presenting signs and symptoms include bleeding such as petechiae, purpura, epistaxis, hemorrhage, and fatigue. Workup should include physical exam, complete blood count, reticulocyte count, and peripheral blood smear. Testing for the presence of antiplatelet antibodies is not generally recommended due to low sensitivity. Bone marrow biopsy is not recommended in the absence of other accompanying cytopenias. However, if a bone marrow biopsy is performed, it would reveal increased megakaryocytes, signifying platelet destruction rather than decreased platelet production (Calvo, 2019).
American Society of Clinical Oncology (ASCO) guidelines for the management of irAEs is based on the grade of thrombocytopenia (see Table 3). Holding the ICI therapy and close monitoring for improvement of the platelet count is recommended. Administration of prednisone (1 mg/kg/day over 2–4 weeks and then tapered over 4–6 weeks) may be considered as well as IVIG (1 g/kg as one-time dose; may be repeated if needed) if rapid increase in platelets is clinically warranted. Rituximab (375 mg/m2 weekly × 4) with or without thrombopoietin receptor agonists are recommended for cases refractory to steroids or IVIG (Brahmer et al., 2018; Lucchini et al., 2019).

Acquired Thrombotic Thrombocytopenic Purpura (TTP)
Acquired thrombotic thrombocytopenic purpura is a thrombotic microangiopathy caused by severely reduced activity of the von Willebrand factor (vWF)-cleaving protease ADAMTS13 (Youssef et al., 2018). This leads to an excess of vWF-causing platelet aggregation, causing small vessel thrombi and organ damage. There are four case reports of TTP due to ICIs (Dickey et al., 2020; King et al., 2017; Lafranchi et al., 2020; Youssef et al., 2018). The classic pentad for the diagnosis of TTP is fever, hemolytic anemia, thrombocytopenia, renal impairment, and neurologic manifestations; however, this pentad may be misleading since it has been identified when TTP is largely fatal due to lack of effective treatment. Therefore, the only reliable factors in current practice are hemolytic anemia and thrombocytopenia without other cause (Youssef et al., 2018). Other clinical manifestations may include abdominal pain, nausea, diarrhea, chest pain, dyspnea, and bleeding/bruising.
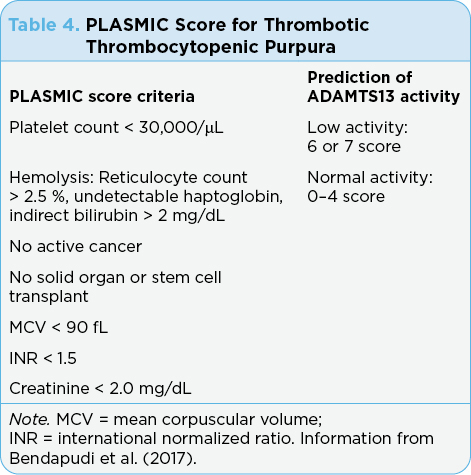
Workup should include physical exam, complete blood count, peripheral blood smear, coagulation studies, urinalysis, blood cultures, and type and screen. Testing may also be sent for ADAMTS13 levels, but this may take several weeks to return. A new scoring tool for TTP called the PLASMIC score (see Table 4) was developed and reported on by Bendapudi and colleagues (2017). ASCO guidelines for the management of irAEs have no grading system for TTP (Brahmer et al., 2018).
Thrombotic thrombocytopenic purpura is a medical emergency requiring rapid intervention (Table 2). Recommended management includes urgent hematology consult and initiation of plasma exchange, as well as IV methylprednisolone
(1 g daily for 3 days; first dose after plasma exchange; Brahmer et al., 2018). Rituximab has been used to manage TTP and has been considered for front-line therapy. However, there are no clear guidelines about when to use rituximab. Therefore, rituximab may be considered, and patients should also receive supportive care with blood and platelet transfusions as needed (Brahmer et al., 2018; Chen et al., 2017).
Post-Transfusion Purpura (PTP)
Post-transfusion purpura is a rare, delayed reaction to blood transfusions (5–10 days following transfusion) and may be challenging to diagnose and differentiate from other entities causing immune-mediated thrombocytopenia. Generally, it is most reported in multiparous women, but there has been a case report of PTP occurring in a patient who had received ICI therapy (Rafei et al., 2017). It can occur after transfusion of any platelet-containing product (i.e., red blood cells or platelets) and is caused by alloimmunization against platelet antigens that leads to complement fixation of platelets. The most frequently involved antigen is antihuman platelet angiten-1a (HPA-1a), but other antigens have also been noted (HPA-1b, HPA-3a, HPA-3b, HPA-4a, HPA-4b; Rafei et al., 2017; Vu & Leavitt, 2018). In the case report of PTP associated with ICI therapy, the antibodies were noted to be against HPA-4a (Vu & Leavitt, 2018).
Clinical suspicion along with serologic findings are important in making the diagnosis. The presence of alloantibodies to platelet antigens is important in supporting the PTP diagnosis in those where the patient’s platelets lack the noted alloantibodies. Since serologic tests are not always readily available at the time the patient presents with thrombocytopenia, high clinical suspicion should prompt immediate treatment (Rafei et al., 2017).
Treatment of PTP includes IVIG, corticosteroids, or plasmapheresis; however, since there is just one single reported case associated with ICIs, it is unclear if IVIG or steroids should be first-line therapy (Table 2). The ICI may need to be discontinued. It is recommended that in severe cases of thrombocytopenia, a higher dose of IVIG should be used (1 g/kg/day for 2 days; Rafei et al., 2017). Platelet transfusions are not effective; however, if needed, HPA-1a–negative patients should receive blood products that are also HPA-1a negative.
Neutropenia/Agranulocytosis
Neutropenia induced by immunotherapy is far less common than neutropenia caused by cytotoxic chemotherapy. In patients treated for lung cancer, the neutropenia rates varied greatly: 1% with immunotherapy vs. 31% with chemotherapy (Delanoy et al., 2019). Rates up to 5% have been reported in patients treated with immunotherapy for Hodgkin lymphoma (Turgeman et al., 2017). Neutropenia secondary to ipilimumab is more frequently reported compared with single-agent PD-1 or PD-L1, although the rationale is poorly understood since neutrophils do not have CTLA-4 expression (Ban-Hoefen et al., 2016; Barbacki et al., 2018).
Although cases of isolated, asymptomatic grade 3 to 4 neutropenia have been reported, the majority of cases are far more complicated and lead to hospitalization (Turgeman et al., 2017). The risk of infection is correlated with the duration and grade of neutropenia (Akhtari et al., 2009). The risk of mortality is largely from bacterial or fungal infection and can include septic shock (Delanoy et al., 2019). It is essential to rule out viral infections (parvovirus B19, Epstein-Barr virus, and cytomegalovirus) as a cause for severe neutropenia (Akhtari et al., 2009). A bone marrow biopsy can show defects in neutrophil precursors that suggests that neutrophils are targeted in their early development (Turgeman et al., 2017). Erythroid and megakaryocytic lineages are usually not involved, while granulocytic lineage will be hypoplastic (Meti et al., 2018) or absent (Ban-Hoefen et al., 2016). There may be moderate eosinophilia noted (Ban-Hoefen et al., 2016). Antineutrophil antibodies may be detected in serum. Less commonly, lipid-laden macrophages can be seen in the marrow (Akhtari et al., 2009).
There are no reported case studies of recovery of severe neutropenia without intervention (Ban-Hoefen et al., 2016). Supportive management plays a fundamental role in its treatment, often with granulocyte colony-stimulating factor (G-CSF) and empiric antibiotics (Barbacki et al., 2018). While the guidelines (e.g., ASCO, NCCN) do not list the management of neutropenia associated with ICIs, case reports have included the use of filgrastim for the management of this uncommon side effect of ICIs (Boegeholz et al., 2020). Oral or intravenous steroids are used with some success; however, when steroids are unsuccessful, additional treatments such as IVIG, cyclosporine, and rabbit antithymocyte globulin (ATG) are used (Ban-Hoefen et al., 2016; Turgemen et al., 2017).
Bicytopenia/Pancytopenia
Hematologic irAEs usually affect a single cell line; however, there are a few case reports where more than one cell line is affected (i.e., bicytopenia, pancytopenia). The reports of bicytopenia have been with the anti–CTLA-4 and PD-1 inhibitors (Atwal et al., 2017; Inadomi et al., 2016). The majority of these cases previously received chemotherapy agents; therefore, a clear link cannot be made between the checkpoint inhibitor and the cytopenia (Rusquec et al., 2014). Bicytopenia is often associated with other disorders such as AA, MDS, disseminate intravascular coagulation (DIC), and TTP. Pancytopenia has also been reported and is associated with nivolumab (Opdivo) and ipilimumab (Rusquec et al., 2014; Tokumo et al., 2018). Some authors indicate that cytopenias are often late occurrences; however, others report the cytopenias occurring early in treatment (Rusquec et al., 2014; Takahashi et al., 2019).
Complete blood counts need to be monitored, and in the case of grade 3 or grade 4 cytopenias, the ICI should be discontinued and high-dose corticosteroids started. Prior cases have been noted to be resistant to corticosteroids, and IVIG was needed to manage the cytopenias. Therefore, IVIG should be considered as front-line therapy (Atwal et al., 2017; Rusquec et al., 2014).
Aplastic Anemia (AA)
Aplastic anemia is a rare and life-threatening failure of the bone marrow that results in cytopenias of the peripheral blood along with trilineage aplasia of the bone marrow. Severe AA is defined as cellularity of the bone marrow less than 25%, or 25% to 50% with less than 30% hematopoietic cells plus two of the following: (a) neutrophil count < 0.5 × 109/L, (b) platelet count < 20 × 109/L, or (c) reticulocyte count < 20 × 109/L. Almost all sporadic AA appears to be immune mediated. Bone marrow suppression that leads to AA is thought to be related to T-cell activation against early hematopoietic progenitors. There have been a few cases of AA reported from ICIs. Of the three cases reported in the literature, two were on dual PD-1 and CTLA-4 therapy and the third had previously received CTLA-4 therapy as a single agent and was receiving nivolumab at the time of diagnosis of AA (Helgadottir et al., 2017; Rouvinov et al., 2019).
Typical symptoms of AA include fatigue, easy bruising or bleeding, and/or infections, but generally there is a long-standing illness. When AA is suspected, a workup should occur rapidly so that other conditions that may mimic AA can be ruled out. Baseline assessment includes a full history and physical exam, complete blood counts with differential, a blood smear, a reticulocyte count, and a bone marrow aspirate and biopsy along with studies of the bone marrow including cytogenetics and fluorescence in situ hybridization (FISH). There is abnormal blast count and the lacunar spaces are extensively replaced by fatty cells. Clonal abnormalities in AA occur in about 12% of cases. The most common abnormalities include abnormalities of chromosome 7 and trisomy 8. In AA, the marrow is hypocellular with no overt dysplasia (Sangiorgio & Calaminici, 2020). Besides ICI therapy, other potential causes include other medications, autoimmune diseases, family or personal history of inherited bone marrow failure disorder, infections, or nutritional deficiencies (Peslak et al., 2018). Screening for Fanconi anemia should also be completed during the workup of pancytopenia. The potential diagnosis of dyskeratosis congenita, hepatitis-associated AA, MDS, or paroxysmal nocturnal hemoglobinuria (PNH) should also be ruled out.
Aplastic anemia can be life threatening; therefore, it is important to detect low peripheral blood counts early in patients receiving ICIs and to withhold ICI and initiate therapy as soon as possible (Helgadottir et al., 2017). Once the diagnosis of AA is made, the treatment is determined by its severity. Patients should be provided aggressive supportive care. Transfusion targets of hemoglobin > 7 g/dL, platelets > 10,000 uL are preferred. The blood products should be irradiated to prevent transfusion-associated graft-vs.-host disease (GVHD). In patients with severe neutropenia, antifungal prophylaxis (e.g., voriconazole or posaconazole) should be used to prevent invasive infections (e.g., aspergillus). In addition, Pneumocystis jiroveci pneumonia prophylaxis should be used during lymphopenia that occurs following ATG therapy (agent other than trimethoprim-sulfamethoxazole since it can cause myelosuppression), such as dapsone, dapsone plus pyrimethamine, or atovaquone. Granulocyte colony-stimulating factor is only used during episodes of febrile neutropenia since no improvement in overall survival has been noted when used in the absence of fever. Antimicrobial prophylaxis with quinolone antibiotics in very severe AA should be administered to reduce the risk of gram-negative sepsis. The routine use of prophylactic antibacterial agents in patients with higher neutrophil counts is not advised (Peslak et al., 2018).
Ultimate treatment of AA is based on age (over or under 40 years), where older adults receive immunosuppressive therapy (IST) while younger adults and children receive bone marrow transplant as the treatment of choice. Immunosuppressive treatment with horse ATG and cyclosporine A (CsA) is recommended in the front line. The optimal duration to administer CsA is not clear, but there is a high rate of relapse when CsA is discontinued early, and maintenance CsA can delay relapses (Peslak et al., 2018). No other treatments have been found to be superior to standard IST (Peslak et al., 2018). Most patients who relapse after having an initial response to IST can be salvaged with full-dose CsA monotherapy and/or a second course of IST with rabbit ATG and CsA or transplant. Other second- and third-line treatment options for refractory AA include danazol, alemtuzumab, androgens, and cyclophosphamide (Peslak et al., 2018).
While there are fairly defined guidelines for the front-line management of severe AA and very severe AA, the management of nonsevere AA is not clearly defined. Expert AA guidelines recommend treating nonsevere AA if patients are transfusion dependent or have neutropenia. Eltrombopag (Promacta) has been proposed as a potential option for nonsevere AA because of its excellent tolerability and efficacy (Peslak et al., 2018).
Myelodysplastic Syndrome (MDS)
Progression of subclinical MDS has been reported in two cases receiving an ICI. The first reported patient was on a clinical trial receiving pembrolizumab (Keytruda) for advanced melanoma (Greenplate et al., 2016). Prior to the initiation of immunotherapy, the patient had stable mild thrombocytopenia with no peripheral blasts, most likely associated with her prior therapy for breast cancer. Over the first 6 months of ICI therapy, the patient developed peripheral blasts, anemia, and progressive thrombocytopenia, prompting bone marrow biopsy confirming refractory anemia with excess blasts. These authors (Greenplate et al., 2016) reported that the peripheral blasts were largely negative for PD-1 and PD-L1 protein expression. They also noted that is it unclear whether pembrolizumab contributed to the progression of MDS in this patient and questioned if the immune system played a role in the development of MDS. The patient initiated decitabine with stabilization of her peripheral blast count and was able to continue on pembrolizumab.
The second reported case had recurrent Hodgkin lymphoma who had multiple prior therapies, including an autologous transplant, and developed grade 3 MDS (Ansell et al., 2015). This patient’s treatment with the ICI was discontinued. The patient’s additional treatment and outcome were not described in the article.
The role of the ICI in the development of MDS is further clouded due to newer data that demonstrated activity of the ICIs in the management of MDS and acute myeloid leukemia. In a study by Garcia-Manera and colleagues (2016), preliminary results demonstrated that PD-1 blockade in combination with azacitidine in patients with untreated higher-risk MDS had clinical activity. In addition, single-agent ipilimumab demonstrated responses in patients with previously treated MDS. However, single-agent nivolumab did not show clinical activity. Additional studies using the ICI in the management of MDS in the front-line and recurrent settings are underway (Chokr et al., 2018).
Cryoglobulinemia
Case studies have reported the occurrence of cryoglobulinemia associated with checkpoint inhibitors (Pellegrino et al., 2017). Two case studies reporting cryoglobulinemia have been noted in the literature: one isolated case and one with possible Sjögren syndrome (Le Burel et al., 2017; Pellegrino et al., 2017). Presentation may include arthralgias, fatigue, and acrocyanosis and necrosis. In the isolated case, cryoglobulins were detected in the blood and was negative after 26 days of corticosteroid therapy (Le Burel et al., 2017; Pellegrino et al., 2017).
Hemophagocytic Lymphohistiocytosis/Cytokine Release Syndrome
An overstimulated immune response may be clinically beneficial; however, it may also lead to hyperinflammation and immune-mediated organ damage. The main cause of hemophagocytic lymphohistiocytosis (HLH) is persistent stimulation of cytotoxic T lymphocytes and natural killer (NK) cells. The persistent stimulation leads to systemic inflammation caused by cytokine storm. Cytokine release syndrome is a collection of inflammatory symptoms that may range from flu-like syndrome to severe vascular leaks and coagulopathy that may lead to organ damage, failure, and death. Presentation of HLH may include fever, splenomegaly, cytopenias (must affect 2 of 3 cell lineages), hypertriglyceridemia and/or hypofibrinogenemia, hemophagocytosis in bone marrow, liver, spleen, or lymph nodes, low or absent NK-cell activity, and hyperferritinemia. The diagnosis is often missed due to lack of awareness. Treatment includes supportive care, immunomodulation, and cytotoxic control (corticosteroids, cyclosporine, methotrexate, etoposide, and IVIG therapy; Sadaat & Jang, 2018).
Eosinophilia
Increased eosinophils in the peripheral blood and tumor-associated tissue eosinophilia are frequently associated with some tumor types, especially in those of epithelial origin such as colon and breast cancer (Moreira et al., 2017). Tumor-associated eosinophilia has been noted to indicate good prognosis in several tumor types such as gastrointestinal cancer; however, it is unclear if it impacts the prognosis of other tumors (i.e., Hodgkin lymphoma) (Davis & Rothenberg, 2014; Gatault et al., 2012). Increased eosinophils have also been noted after treatment with immunotherapy such as IL-2, IL-4, GM-CSF and anti–CTLA-4 antibody (Davis & Rothenberg, 2014; Gatault et al., 2012).
Overall, 3% to 37% of patients receiving ICI develop increased eosinophil counts (Bernard-Tessier et al., 2017; Moreira et al., 2017). Researchers have reported eosinophilia (absolute eosinophil count greater than 0.5 g/L) occurring in 27.1% of patients receiving PD-1 or PD-L1 inhibitors, but noted that about half of those patients had eosinophilia since the time of starting therapy (Kizawa et al., 2019). There have been reports of increased eosinophil counts in the peripheral blood of patients who have received ipilimumab and its association with improved survival (Delyon et al., 2013; Umansky et al., 2016). The increased eosinophil count, which occurs following treatment with ipilimumab or pembrolizumab, has been noted to impact survival. However, increased eosinophilia noted prior to starting treatment has not been found to affect survival (Weide et al., 2016). The median time to increase in eosinophil count is 3 months, with peak eosinophil count of 1,000/mm3 noted at a median of 6.4 months (Yang et al., 2019). Kizawa and colleagues (2019) indicated a relationship between the presence of eosinophilia and subsequent irAEs. The most common irAEs associated with eosinophilia were endocrine disorders such as thyroid dysfunction and adrenal insufficiency and skin disorders.
There are other disorders of the eosinophils that have been reported to be associated with checkpoint inhibitors. One of these is eosinophilic fasciitis that is a fibrosing disorder that causes inflammatory infiltration of the subcutaneous fascia. An increase in the eosinophil count is not needed for the diagnosis. This disorder is managed with corticosteroids (Chan et al., 2019).
An additional irAE that has been noted to be associated with checkpoint inhibitors includes the DRESS syndrome (Drug Reaction with Eosinophilia and Systemic Symptoms). Characteristics include the presence of eosinophilia, rash, and involvement of the internal organs. DRESS syndrome needs to be detected early and managed quickly (Lu et al., 2019).
A third rare disorder associated with elevated eosinophils that has been reported to be associated with checkpoint inhibitors is eosinophilic granulomatosis with polyangiitis (also known as Churg-Strauss syndrome). It is characterized by necrotizing vasculitis of small- and medium-sized systemic blood vessels. It can be distinguished from other disorders by the association of asthma, rhinosinusitis, and peripheral eosinophilia (Chakraborty & Aeddula, 2021). In this review, only a case study can be found regarding eosinophilic granulomatosis with polyangiitis in a patient receiving nivolumab (Roger et al., 2019). In that patient, there was no evidence of vasculitis, but the patient did have asthma so the true diagnosis in this situation is unclear. Of note is that in this patient, a complete response persisted 12 months after the discontinuation of nivolumab.
Discussion
The use of ICIs has had a significant impact on the management of malignancies. These agents are associated with unique AEs that most commonly include pneumonitis, colitis, and dermatitis. Hematologic AEs associated with ICIs are rare but have varying severity. The hematologic AEs associated with ICIs that have been reported can affect each of the cell lines leading to neutropenia, thrombocytopenia, and anemia. Besides the suppression of each of the cell lines, other hematologic AEs include cryoglobulinemia, hemophagocytic lymphocytosis, and eosinophilia. The impact on the blood counts is usually related to prior chemotherapy treatments; however, health-care providers need to be cognizant of the potential impact of ICIs on blood counts.
Typical treatment parameters, such as holding treatment for an absolute neutrophil count of less than 1,500, may not be appropriate in patients receiving ICIs. The electronic medical record order sets need to include parameters (for holding treatment) that are meaningful for patients receiving ICIs. While the majority of the AEs that impact the hematologic system are managed with steroids, other treatments may be needed, such as IVIG or apheresis. It is important for advanced practitioners to understand the potential for these AEs so patients can receive prompt treatment.
Disclosure
Ms. Rogers has served on speakers bureaus for Bristol Myers Squibb, Seattle Genetics, Teva, Genentech, and AbbVie. The remaining authors have no conflicts of interest to disclose.
References
Akhtari, M., Waller, E., Jaye, D., Lawson, D., Ibrahim, R., Papadopoulos, N., & Arellano, M. (2009). Neutropenia in a patient treated with ipilimumab (anti-CTLA-4 antibody). Journal of Immunotherapy, 32(3), 322–324. https://doi.org/10.1097/cji.0b013e31819aa40b
Ansell, S., Lesokhin, A., Borrello, I., Halwani, A., Scott, E., Gutierrez, M.,...Armand, P. (2015). PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. New England Journal of Medicine, 372, 311–319. https://doi.org/10.1056/NEJMoa1411087
Atwal, D., Joshi, K., Ravilla, R., & Mahmoud, F. (2017). Pembrolizumab-induced pancytopenia: A case report. Permanente Journal, 21. https://doi.org/19.7812/TPP/17-004
Ban-Hoefen, M., Burack, R., Lievert, L., & Sahasrabudhe, D. (2016). Ipilimumab-induced neutropenia in melanoma. Journal of Investigative Medicine, 1–5. https://doi.org/10.1177/2324709616661835
Barbacki, A., Maliha, P., Hudson, M., & Small, D. (2018). A case of severe pembrolizumab-induced neutropenia. Anti-Cancer Drugs, 29(8), 817–819. https://doi.org/10.1097/cad.0000000000000661
Bendapudi, P., Hurwitz, S., Fry, A., Marques, M., Waldo, S., Li, A.,...Makar, R. (2017). Derivation and external validation of the PLASMIC score for rapid assessment of adults with thrombotic microangiopathies: A cohort study. Lancet Haematology, 4(4), e157–e164. https://doi.org/10.1016/s2352-3026(17)30026-1
Berentsen, S., Randen, U., & Tjonnfjord, G. (2015). Cold agglutinin-mediated autoimmune hemolytic anemia. Hematology Oncology Clinics of North America, 29(3), 455–471. http://dx.doi.org/10.1016/j.hoc.2015.01.002
Bernard-Tessier, A., Jeanville, P., Champiat, S., Lazarovici, J., Voisin, A., Mateus, C.,...Michot, J. (2017). Immune-related eosinophilia induced by anti-programmed death 1 or death-ligand 1 antibodies. European Journal of Cancer, 81, 135–137. https://doi.org/10.1016/j.ejca.2017.05.017
Boegeholz, J., Brueggen, C., Pauli, C., Dimitriou, F., Haralambieva, E., Dummer, R.,...Widmer, C. (2020). Challenges in diagnosis and management of neutropenia upon exposure to immune-checkpoint inhibitors: Meta-analysis of a rare immune-related adverse side effect. BMC Cancer, 20, Article number: 300. https://doi.org/10.1186/s12885-020-06763-y
Brahmer, J. R., Lacchetti, C., Schneider, B. J., Atkins, M. B., Brassil, K. J., Caterino, J. M.,…Puzanov, I. (2018). Management of Immune-Related Adverse Events in Patients Treated With Immune Checkpoint Inhibitor Therapy: American Society of Clinical Oncology Clinical Practice Guideline. Journal of Clinical Oncology, 36(17), 1714–1768. https://doi.org/10.1200/jco.2017.77.6385
Chakraborty, R., & Aeddula, N. (2021). Churg Strauss Syndrome. Churg Strauss Syndrome - StatPearls - NCBI Bookshelf (nih.gov)
Calvo, R. (2019). Hematological side effects of immune checkpoint inhibitors: The example of immune-related thrombocytopenia. Frontiers of Pharmacology, 10. https://doi.org/10.3389/fphar.2019.00454
Chan, K. K., Magro, C., Shoustari, A., Rudin, C., Rotemberg, V., Rossi, A.,...Bass, A. (2019). Eosinophilic fasciitis following checkpoint inhibitor therapy: Four cases and a review of the literature. Oncologist, 25(2), 140–149. https://doi.org/10.1634/theoncologist.2019-0508
Chen, H., Fu, A., Wang, J., Wu, T., Li, Z., Tang, J.,...Qing, L. (2017). Rituximab as first-line treatment for acquired thrombotic thrombocytopenic purpura. Journal of International Medical Research, 45(3), 1253–1260. https://dx.doi.org/10.1177%2F0300060517695646
Chokr, N., Patel, R., Wattamwar, K., & Chokr, S. (2018). The rising era of immune checkpoint inhibitors in myelodysplastic syndromes. Advances in Hematology, 2018, Article ID 2458679. https://doi.org/10.1155/2018/2458679
Davis, B., & Rothenberg, M. (2014). Eosinophils and cancer. Cancer Immunotherapy Research, 2(1), 1–8. https://doi.org/10.1158/2326-6066.CIR-13-0196
Davis, E., Salem, J., Young, A., Green, J., Ferrell, P., Ancell, K.,...Johnson, D. (2019). Hematologic complications of immune checkpoint inhibitors. Oncologist, 24(5), 584–588. https://doi.org/10.1634/theoncologist.2018-0574
Delanoy, N., Michot, J.-M., Comont, T., Kramkimel, N., Lazarovici, J., Dupont, R.,…Biscay, P. (2019). Haematological immune-related adverse events induced by anti-PD-1 or anti-PD-L1 immunotherapy: A descriptive observational study. Lancet Haematology, 6(1), e48–e57. https://doi.org/10.1016/s2352-3026(18)30175-3
Delyon, J., Mateus, C., Lefeuvre, D., Lanoy, E., Zitvogel, L., Chaput, N.,…Robert, C. (2013). Experience in daily practice with ipilimumab for the treatment of patients with metastatic melanoma: An early increase in lymphocyte and eosinophil counts is associated with improved survival. Annals of Oncology, 24(6), 1697–1703. https://doi.org/10.1093/annonc/mdt027
Dickey, M. S., Raina, A. J., Gilbar, P. J., Wisniowski, B. L., Collins, J. T., Karki, B., & Nguyen, A. D. (2019). Pembrolizumab-induced thrombotic thrombocytopenic purpura. Journal of Oncology Pharmacy Practice, 26(5), 1237–1240. https://doi.org/10.1177/1078155219887212
Garcia-Manero, G., Daver, N. G., Montalban-Bravo, G., Jabbour, E. J., DiNardo, C. D., Kornblau, S. M.,…Yang, H. (2016). A phase II study evaluating the combination of nivolumab (nivo) or ipilimumab (ipi) with azacitidine in pts with previously treated or untreated myelodysplastic syndromes (MDS). Blood, 128(22), 344–344. https://doi.org/10.1182/blood.v128.22.344.344
Gnanapandithan, K., Kharel P., Grimshaw, A., & Giri, S. (2019). Hematologic immune-related adverse events from immune checkpoint inhibitors: A systematic review of case-reports and case-series. Blood (ASH Annual Abstracts), 134(Supplement 1). https://doi.org/10.1182/blood-2019-132203
Gatault, S., Legrand, F., Delbeke, M., Loiseau, S., & Capron, M. (2012). Involvement of eosinophils in the anti-tumor response. Cancer Immunology and Immunotherapy, 61(9), 1527–1534. https://doi.org/10.1007/s00262-012-1288-3
Gordon, H., Wade, T., Chin, K., Dickstein, J., & Gajewski, T. (2009). Immune-mediated red cell aplasia after anti-CTLA-4 immunotherapy for metastatic melanoma. Cancer Immunology and Immunotherapy, 58(8), 1351–1353. https://doi.org/10.1007/s00262-008-0627-x
Greenplate, A., Johnson, D., Roussel, M., Savona, M., Sosman, J., Puzanov, I.,…Irish, J. (2016). Myelodysplastic syndrome revealed by systems immunology in a melanoma patient undergoing anti-PD-1 therapy. Cancer Immunology Research, 4(6), 474–480. https://doi.org/10.1158/2326-6066.cir-15-0213
Hasanov, M., Konoplev, S., & Hernandez, C. (2018). Nivolumab-induced cold agglutinin syndrome successfully treated with rituximab. Blood Advances, 2(15), 1865–1868. https://doi.org/10.1182/bloodadvances.2018019000
Helgadottir, H., Kis, L., Ljungman, P., Larkin, J., Kefford, R., Ascierto, P.,...Masucci, G. (2017). Lethal aplastic anemia caused by dual immune checkpoint blockade in metastatic melanoma. Annals of Oncology, 28(7), 1672–1673. https://doi.org/10.1093/annonc/mdx177
Inadomi, K., Kumagai, H., Arita, S., Tsuruta, N., Takayoshi, K., Mishima, K.,…Baba, E. (2016). Bi-cytopenia possibly induced by anti-PD-1 antibody for primary malignant melanoma of the esophagus. Medicine, 95(29), e4283. https://doi.org/10.1097/md.0000000000004283
Khan, U., Ali, F., Khurram, M. S., Zaka, A., & Hadid, T. (2017). Immunotherapy-associated autoimmune hemolytic anemia. Journal for ImmunoTherapy of Cancer, 5(1), Article number: 15. https://doi.org/10.1186/s40425-017-0214-9
King, J., de la Cruz, J., & Lutzky, J. (2017). Ipilimumab-induced thrombotic thrombocytopenic purpura (TTP). Journal for ImmunoTherapy of Cancer, 5(1). https://doi.org/10.1186/s40425-017-0224-7
Kizawa, R., Miura, Y., Oda, Y., Nagaoka, Y., Ozaki, Y., Kondoh, C.,...Masuda, J. (2019). Eosinophilia during treatment of immune checkpoint inhibitors (ICIs) to predict succeeding onset of immune-related adverse events (irAEs) [Abstract e14110]. Journal of Clinical Oncology (ASCO Annual Meeting Abstracts), 37(15). https://doi.org/10.1200/JCO.2019.37.15_suppl.e14110
Kong, B., Micklethwaite, K., Swaminathan, S., Kefford, R., & Carlino, M. (2016). Autoimmune hemolytic anemia induced by anti-PD-1 therapy in metastatic melanoma. Melanoma Research, 26(2), 202–204. https://doi.org/10.1097/CMR.0000000000000232
Lafranchi, A., Springe, D., Rupp, A., Ebnöther, L., & Zschiedrich, S. (2020). Thrombotic thrombocytopenic purpura associated to dual checkpoint inhibitor therapy for metastatic melanoma. CEN Case Reports, 9(3), 289–290. https://doi.org/10.1007/s13730-020-00454-0
Le Burel, S., Champiat, S., Mateus, C., Marabelle, A., Michot, J., Robert, C.,...Lambotte, O. (2017). Prevalence of immune-related systemic adverse events in patients treated with anti-programmed cell death 1/anti-programmed cell death-ligand 1 agents: A single-centre pharmacovigilance database analysis. European Journal of Cancer, 82, 34–44. https://doi.org/10.1016/j.ejca.2017.05.032
Leaf, R., Ferreri, C., Rangachari, D., Mier, J., Witteles, W., Ansstas, G.,...Leaf, D. (2019). Clinical and laboratory features of autoimmune hemolytic anemia associated with immune checkpoint inhibitors. American Journal of Hematology, 94(5), 563–574. https://doi.org/10.1002/ajh.25448
Lu, J., Thuraisingam, T., Chergui, M., & Nguyen, K. (2019). Nivolumab-associated DRESS syndrome: A case report. Journal of the American Academy of Dermatology, 5(3), 216–218. https://dx.doi.org/10.1016%2Fj.jdcr.2018.11.017
Lucchini, E., Zaja, F., & Bussel, J. (2019). Rituximab in the treatment of immune thrombocytopenia: What is the role of this agent in 2019? Haematologica, 104(6), 1124–1135. https://doi.org/10.3324/haematol.2019.218883
Meti, N., Petrogiannis-Haliotis, T., & Esfahani, K. (2018). Refractory neutropenia secondary to dual immune checkpoint inhibitors that required second-line immunosuppression. Journal of Oncology Practice, 14(8), 514–516. https://doi.org/10.1200/JOP.18.00177
Michot, J., Lazarovici, J., Tieu, A., Champiat, S., Voisin, A., Ebbo, M.,...Lambotte, O. (2019). Haematological immune-related adverse events with immune checkpoint inhibitors, how to manage? European Journal of Cancer, 122, 72–90. https://doi.org/10.1016/j.ejca.2019.07.014
Moreira, A., Leisgang, W., Schuler, G., & Heinzerling, L. (2017). Eosinophilic count as a biomarker for prognosis of melanoma patient and its importance in the response to immunotherapy. Immunotherapy, 9(2), 115–121. https://doi.org/10.2217/imt-2016-0138
National Cancer Institute. (2006). Common Terminology Criteria for Adverse Events (CTCAE) v3.0. https://ctep.cancer.gov/protocoldevelopment/electronic_applications/ctc.htm
Noseda, R., Bertoli, R., Muller, L., & Ceschi, A. (2019). Haemophagocytic lymphohistiocytosis in patients treated with immune checkpoint inhibitors: Analysis of WHO global database of individual case safety reports. Journal for ImmunoTherapy of Cancer, 7, 117. https://doi.org/10.1186/s40425-019-0598-9
Ogawa, K., Ito, J., Fujimoto, D., Morita, M., Yoshizumi, Y., Ariyoshi, K.,...Katakami, N. (2018). Exacerbation of autoimmune hemolytic anemia induced by the first dose of programmed death-1 inhibitor pembrolizumab: A case report. Investigational New Drugs, 36(3), 509–512. https://doi.org/10.1007/s10637-018-0561-5
Palla, A., Kennedy, D., Mosharrf, H., & Doll, D. (2016). Autoimmune hemolytic anemia as a complication of nivolumab therapy. Case Reports in Oncology, 9(3), 691–697. https://doi.org/10.1159/000452296
Pellegrino, B., Musolino, A., & Tiseo, M. (2017). Anti-PD-1-related cryoglobulinemia with nivolumab in NSCLC patient. Annals of Oncology, 28(6), 1405–1406. https://doi.org/10.1093/annonc/mdx126
Peslak, S., Olson, T., & Babushok, D. (2018). Diagnosis and treatment of aplastic anemia. Current Treatment Options in Oncology, 18, Article number: 70. https://doi.org/10.1007/s11864-017-0511-z
Rafei, H., Yunus, R., & Nassereddine, S. (2017). Post-transfusion purpura: A case report of an underdiagnosed phenomenon. Cereus, 9(5), e1207. https://doi.org/10.7759/cureus.1207
Roger, A., Groh, M., Lorillon, G., Le Pendu, C., Maillet, J., Arangalage, D.,...Delyon, J. (2019). Eosinophilic granulomatosis with polyangitis (Churg-Strauss) induced by immune checkpoint inhibitors. Annals of the Rheumatic Diseases, 78, e82. https://ard.bmj.com/content/78/8/e82.info
Roumier, M., Loustau, V., Guillaud, C., Languille, L., Mahevas, M., Khellaf, M.,...Michel, M. (2014). Characteristics and outcome of warm autoimmune hemolytic anemia in adults: New insights based on a single-center experience of 60 patients. American Journal of Hematology, 89(9), E150–E155. https://doi.org/10.1002/ajh.23767
Rouvinov, K., Nalbandyan, K., Kozlov, V., Peled, N., & Yakobson, A. (2019). Nivolumab induced lethal aplastic anemia in a patient with metastatic melanoma. Case Reports in Oncology, 12, 29–32. https://doi.org/10.1159/000495980
Rusquec, P., Saint-Jean, M., Brocard, A., Peuvrel, L., Khammari, A., Quereux, G. & Dreno, B. (2014). Ipilimumab-induced autoimmune pancytopenia in a case of metastatic melanoma. Journal of Immunotherapy, 37(6), 348–350. https://doi.org/10.1097/cji.0000000000000041
Sadaat, M., & Jang, S. (2018). Hemophagocytic lymphohistiocytosis with immunotherapy: Brief review and case report. Journal for ImmunoTherapy of Cancer, 6. https://doi.org/10.1186/s40425-018-0365-3
Sangiorgio, V., & Calaminici, M. (2020). Bone marrow nonneoplastic alterations in cellularity-aplastic anemia. Pathology Outlines. http://www.pathologyoutlines.com/topic/bonemarrowaplasticanemia.html.
Shiuan, E., Beckermann, K. E., Ozgun, A., Kelly, C., McKean, M., McQuade, J.,...Johnson, D. (2017). Journal of Immunotherapy of Cancer, 5. https://doi.org/10.1186/s40425-017-0210-0
Sui, J. D., Wang, Y., Wan, Y., & Wu, Y. Z. (2018). Risk of hematologic toxicities with programmed cell death-1 inhibitors in cancer patients: A meta-analysis of current studies. Drug Design, Development and Therapy, 2018(12), 1645–1657. https://doi.org/10.2147/DDDT.S167077
Takahashi, A., Kubo, A., Mizuno, S., Kasai, K., Asi, N., Yonezawa, T.,...Yamaguchi, E. (2019). Bicytopenia in primary lung melanoma treated with nivolumab. Internal Medicine, 58(6), 827–831. https://doi.org/10.2169/internalmedicine.1011-18
Tanios, G., Doley, P., & Munker, R. (2018). Autoimmune hemolytic anemia associated with the use of immune checkpoint inhibitors for cancer: 68 cases from the Food and Drug Administration database and review. European Journal of Haematology, 102(2), 157–152. https://doi.org/10.1111/ejh.13187
Tokumo, K., Masuda, T., Miyama, T., Miura, S., Yamaguchi, K., Sakamoto, S.,...Hattori, N. (2018). Nivolumab-induced severe pancytopenia in a patient with lung adenocarcinoma. Lung Cancer, 119, 21–24. https://doi.org/10.1016/j.lungcan.2018.02.018
Turgeman, I., Wollner, M., Hassoun, G., Bonstein, L., & Bar-Sela, G. (2017). Severe complicated neutropenia in two patients with metastatic non-small cell lung cancer treated with nivolumab. Anticancer Drugs, 28(7), 811–814. https://doi.org/10.1097/CAD.0000000000000520
Umansky, V., Utikal, J., & Gebhardt, C. (2016). Predictive immune markers in advanced melanoma patients treated with ipilimumab. Oncoimmunology, 5(6), e1158901. https://doi.org/10.1080/2162402X.2016.1158901
Vu, K., & Leavitt, A. (2018). Post-transfusion purpura wit antibodies against human platelet antigen-4a following checkpoint inhibitor therapy: A case report and review of the literature. Transfusion, 58(10), 2265–2269. https://doi.org/10.1111/trf.14824
Weide, B., Martens, A., Hassel, J., Berking, C., Postow, M., Bisschop, K.,...Wolchok, J. (2016). Baseline biomarkers for outcome of melanoma patients treated with pembrolizumab. Clinical Cancer Research, 22(22). https://doi.org/10.1158/1078-0432.ccr-16-0127
Yang, J., Lagana, S., Saenger, Y., & Carvajal, R. (2019). Dual checkpoint inhibitor-associated eosinophilic enteritis. Journal for ImmunoTherapy of Cancer, 7, 310. https://doi.org/10.1186/s40425-019-0743-5
Youssef, A., Kasso, N., Torloni, A. S., Stanek, M., Dragovich, T., Gimbel, M., & Mahmoud, F. (2018). Thrombotic thrombocytopenic purpura due to checkpoint inhibitors. Case Reports in Hematology, 2018, Article ID 2464619. https://doi.org/10.1155/2018/2464619
Yuki, A., Takenouchi, T., Takatsuka, S., & Ishiguro, T. (2017). A case of pure red cell aplasia during nivolumab therapy for cardiac metastatic melanoma. Melanoma Research, 27(6), 635–637. https://doi.org/10.1097/cmr.0000000000000392